
New Bufadienolides Isolated from the Roots of Kalanchoe daigremontiana (Crassulaceae)



Abstract
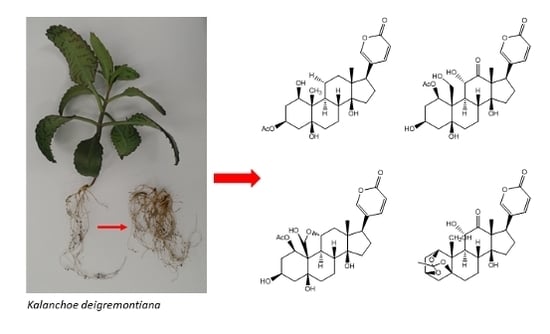
:
1. Introduction
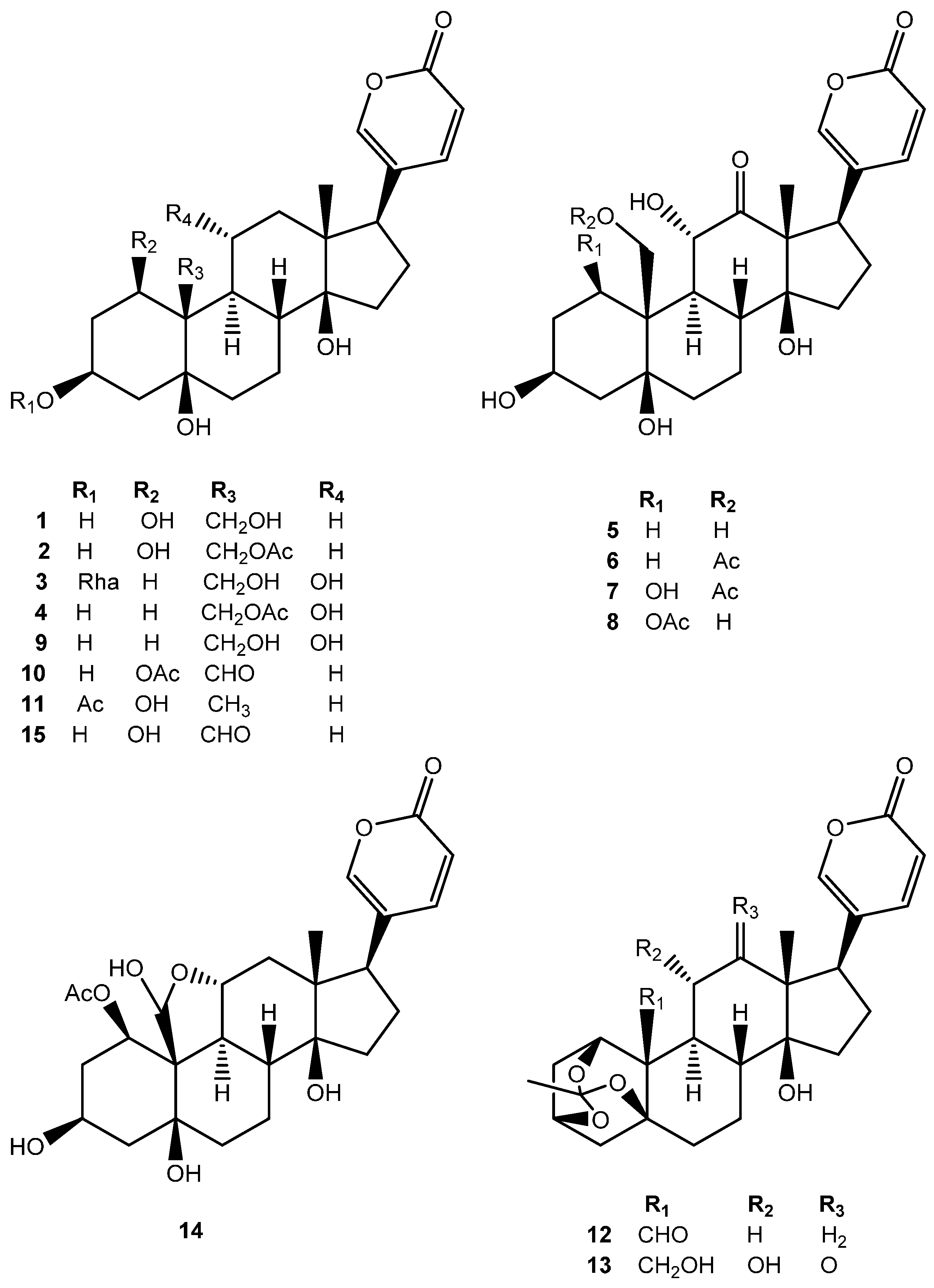
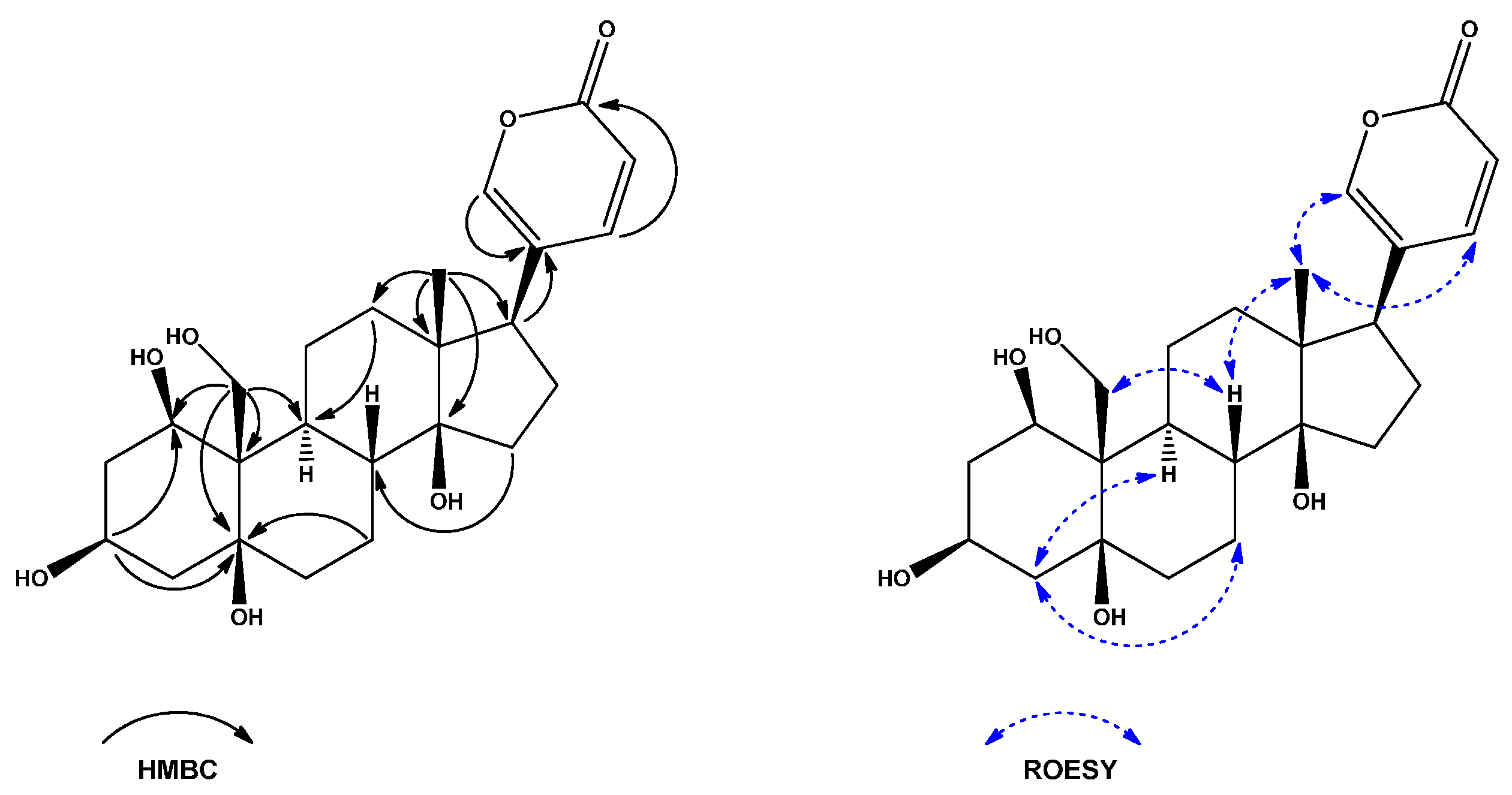
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Acid Hydrolysis of Compound 3 and Determination of the Absolute Configuration of the Sugar
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Herrera, I.; Nassar, J.M. Reproductive and recruitment traits as indicators of the invasive potential of Kalanchoe daigremontiana (Crassulaceae) and Stapelia gigantean (Apocynaceae) in a neotropical arid zone. J. Arid Environ. 2009, 73, 978–986. [Google Scholar] [CrossRef]
- Anisimov, M.M.; Gerasimenko, E.L.; Chaikina, E.L.; Serebryakov, Y.M. Biological activity of metabolites of the herb Kalanchoe daigremontiana (Hamet de la Bathie) Jacobs et Perr. Biol. Bull. 2009, 36, 568–574. [Google Scholar] [CrossRef]
- Misra, S.B.; Dixit, S.N. Antifungal activity of leaf extracts of some higher plants. Acta Bot. Indica 1979, 7, 147–150. [Google Scholar]
- Nassis, C.Z.; Haebisch, E.M.A.B.; Giesbrecht, A.M. Antihistamine activity of Bryophillum calycinum. Braz. J. Med. Res. 1992, 25, 929–936. [Google Scholar]
- Moraes, V.L.G.; Santos, L.F.M.; Castro, S.B.; Loureiro, L.H.; Lima, O.A.; Souza, M.L.M.; Yien, L.M.K.; Rossi-Bergmann, B.; Costa, S.S. Inhibition of lymphocyte activation by extracts and fractions of Kalanchoe, Alternanthera, Paullinea, and Mikania species. Phytomedicine 1994, 1, 199–204. [Google Scholar] [CrossRef]
- Rossi-Bergmann, B.; Costa, S.S.; Borges, M.B.S.; da Silva, S.A.; Noleto, G.R.; Souza, M.L.M.; Moraes, V.L.G. Immunosuppressive effect of the aqueous extract of Kalanchoe pinnata in mice. Phytother. Res. 1994, 8, 399–402. [Google Scholar] [CrossRef]
- Da-Silva, S.A.G.; Costa, S.S.; Mendonca, S.C.F.; Silva, E.M.; Moraes, V.L.G.; Rossi-Bergmann, B. Therapeutic effect of oral Kalanchoe pinnata leaf extract in murine leishmaniasis. Acta Trop. 1995, 60, 201–210. [Google Scholar] [CrossRef]
- Da-Silva, S.A.G.; Costa, S.S.; Rossi-Bergmann, B. The anti-leishmanial effect of Kalanchoe is mediated by nitric oxide intermediates. Parasitology 1999, 118, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Supratman, U.; Fujita, T.; Akiyama, K.; Hayashi, H. New insecticidal bufadienolide, bryophyllin C, from Kalanchoe pinnata. Biosci. Biotechnol. Biochem. 2000, 64, 1309–1311. [Google Scholar] [CrossRef]
- Supratman, U.; Fujita, T.; Akiyama, K.; Hayashi, H. Insecticidal compounds from Kalanchoe daigremontiana × tubiflora. Phytochemistry 2001, 58, 311–314. [Google Scholar] [CrossRef]
- Supratman, U.; Fujita, T.; Akiyama, K.; Hayashi, H.; Murakami, A.; Sakai, H.; Koshimizu, K.; Ohigashi, H. Anti-tumor promoting activity of bufadienolides from Kalanchoe pinnata and K. daigremontiana × tubiflora. Biosci. Biotechnol. Biochem. 2001, 65, 947–949. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.P.; Dixit, V.K. Hepatoprotective activity of leaves of Kalanchoe pinnata Pers. J. Ethnopharmacol. 2003, 86, 197–202. [Google Scholar] [CrossRef]
- Costa, S.S.; de Lourdes, M.; de Souza, M.L.M.; Ibrahim, T.; Oliveira de Melo, G.M.; de Almeida, A.P.; Guette, C.; Férézou, J.P.; Koatz, V.L.G. Kalanchosine dimalate, an anti-inflammatory salt from Kalanchoe brasiliensis. J. Nat. Prod. 2006, 69, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.R.; Ho, Y.L.; Huang, S.C.; Huang, T.H.; Lai, S.C.; Tsai, J.C.; Wang, C.Y.; Huang, G.J.; Chang, Y.S. Antioxidant, anti-inflammatory and antiproliferative activities of Kalanchoe gracilis (L.) dc stem. Am. J. Chin. Med. 2011, 39, 1275–1290. [Google Scholar] [CrossRef] [PubMed]
- Cruz, E.A.; Reuter, S.; Martin, H.; Dehzad, N.; Muzitano, M.F.; Costa, S.S.; Rossi-Bergmann, B.; Buhl, R.; Stassen, M.; Taube, C. Kalanchoe pinnata inhibits mast cell activation and prevents allergic airway disease. Phytomedicine 2012, 19, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Krenn, L.; Kopp, B. Bufadienolides from animal and plant sources. Phytochemistry 1998, 48, 1–29. [Google Scholar] [CrossRef]
- Tian, H.-Y.; Wang, L.; Zhang, X.-Q.; Zhang, D.-M.; Wang, Y.; Liu, J.-S.; Jiang, R.-W.; Ye, W.-C. New bufadienolides and C23 steroids from the venom of Bufo bufo gargarizans. Steroids 2010, 75, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Shen, A.; Wang, C.; Yan, J.; Zhao, W.; Liang, X. Efficient purification of active bufadienolides by a class separation method based on hydrophilic solid-phase extraction and reversed-phase high performance liquid chromatography. J. Pharm. Biomed. Anal. 2014, 97, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Green, B.; Snatzke, F.; Snatzke, G.; Pettit, G.R.; Kamano, Y.; Niven, M.L. Circular Dichroism of Bufadienolides. Croat. Chem. Acta 1985, 58, 371–387. [Google Scholar]
- Goetz, M.A.; Wiemer, D.F.; Haynes, L.W.; Meinwald, J.; Eisner, T. Lucibufagins. Part III. 11-Oxo-and 12-oxo-bufalins, defensive steroids from the fireflies Photinus ignitus and P. marginellus (Coleoptera: Lampyrida). Helv. Chim. Acta 1979, 62, 1396–1400. [Google Scholar] [CrossRef]
- González, A.; Schroeder, F.C.; Attygale, A.B.; Svatoš, A.; Meinwald, J.; Eisner, T. Metabolic transformations of acquired lucibufagins by firefly “femmes fatales”. Chemoecology 1999, 9, 105–112. [Google Scholar] [CrossRef]
- Wagner, H.; Lotter, H.; Fischer, M. The toxic and sedative bufadienolides of Kalanchoe daigremontiana Hamet et Perr. Helv. Chim. Acta 1986, 69, 359–367. [Google Scholar] [CrossRef]
- Żuchowski, J.; Pecio, Ł.; Stochmal, A. Novel Flavonol Glycosides from the Aerial Parts of Lentil (Lens culinaris). Molecules 2014, 19, 18152–18178. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.J.; Simonet, A.M.; Calle, J.M.; Pecio, Ł.; Guerra, J.O.; Stochmal, A.; Macías, F.A. Phytotoxic steroidal saponins from Agave offoyana leaves. Phytochemistry 2014, 105, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.



{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| 1 | 71.7 | 69.9 | 22.4 | 22.1 | 21.9 | 22.0 | 71.0 | 74.1 |
| 2 | 34.6 | 34.3 | 27.4 | 28.8 | 29.0 | 28.6 | 34.6 | 32.1 |
| 3 | 68.1 | 67.9 | 76.0 | 68.6 | 68.9 | 68.4 | 67.9 | 67.8 |
| 4 | 39.3 | 39.5 | 36.4 | 38.6 | 38.4 | 38.5 | 40.0 | 38.9 |
| 5 | 76.5 | 75.3 | 77.5 | 75.9 | 78.1 | 75.9 | 76.1 | 76.5 |
| 6 | 37.1 | 36.9 | 36.2 | 36.8 | 36.5 | 36.6 | 37.0 | 36.5 |
| 7 | 24.6 | 24.6 | 24.7 | 24.8 | 24.8 | 24.5 | 24.6 | 24.6 |
| 8 | 42.5 | 42.6 | 41.2 | 41.5 | 39.8 | 40.1 | 40.1 | 39.8 |
| 9 | 42.5 | 42.2 | 45.7 | 45.4 | 43.9 | 43.6 | 46.3 | 46.5 |
| 10 | 47.0 | 47.0 | 45.3 | 45.7 | 45.4 | 46.1 | 49.3 | 49.3 |
| 11 | 23.7 | 24.0 | 69.6 | 69.8 | 75.1 | 75.5 | 74.9 | 74.9 |
| 12 | 42.1 | 42.0 | 51.6 | 52.0 | 214.5 | 214.2 | 213.9 | 214.2 |
| 13 | 49.6 | 49.6 | 50.1 | 50.1 | 63.6 | 63.6 | 63.4 | 63.4 |
| 14 | 85.9 | 85.9 | 85.5 | 85.4 | 85.6 | 85.6 | 85.4 | 85.4 |
| 15 | 33.1 | 33.1 | 33.1 | 32.9 | 32.7 | 32.7 | 32.9 | 32.9 |
| 16 | 29.8 | 29.7 | 29.6 | 29.6 | 29.0 | 29.0 | 28.9 | 28.8 |
| 17 | 52.1 | 52.1 | 51.9 | 51.9 | 42.1 | 42.0 | 42.1 | 42.1 |
| 18 | 17.3 | 17.3 | 18.4 | 18.2 | 17.9 | 18.0 | 17.7 | 17.5 |
| 19 | 62.7 | 63.9 | 65.3 | 67.8 | 65.9 | 67.5 | 64.3 | 61.2 |
| 20 | 125.0 | 124.9 | 124.4 | 124.5 | 123.1 | 123.0 | 123.0 | 123.1 |
| 21 | 150.5 | 150.5 | 150.6 | 150.7 | 151.6 | 151.6 | 151.7 | 151.7 |
| 22 | 149.3 | 149.3 | 149.1 | 149.1 | 149.1 | 149.0 | 149.0 | 149.1 |
| 23 | 115.5 | 115.5 | 115.5 | 115.5 | 115.9 | 115.9 | 115.9 | 115.8 |
| 24 | 164.8 | 164.7 | 164.7 | 164.7 | 164.4 | 164.4 | 164.4 | 164.4 |
| 1-COCH3 | 172.6 | |||||||
| 1-COCH3 | 21.8 | |||||||
| 19-COCH3 | 173.0 | 173.0 | 172.8 | 173.1 | ||||
| 19-COCH3 | 21.3 | 21.2 | 21.2 | 21.2 | ||||
| Rha | ||||||||
| 1′ | 100.9 | |||||||
| 2′ | 72.6 | |||||||
| 3′ | 72.5 | |||||||
| 4′ | 73.8 | |||||||
| 5′ | 70.6 | |||||||
| 6′ | 18.0 |
| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1α | 4.42, dd (3.3, 3.3) | 4.36, dd (3.3, 3.3) | 2.41, br d (13.6) | 2.50, ddd (13.8, 3.0, 3.0) |
| 1β | 2.03, ddd (13.6, 13.6, 2.5) | 1.73, ddd (13.9, 13.9, 3.5) | ||
| 2α | 1.98, ddd (14.9, 3.5, 3.5) | 2.00, ddd (15.1, 2.7, 2.7) | 1.96, ddd (14.1, 2.6, 2.6) | 1.89, dddd (13.8, 13.8, 3.0, 3.0) |
| 2β | 2.08, dddd (15.0, 2.5, 2.5, 2.5) | 2.09, dd (15.2, 1.5) | 1.80, br d (15.0) | 1.63, dddd (14.5, 3.3, 3.0, 3.0) |
| 3α | 4.22, br s | 4.26, br s | 4.15, br s | 4.14, dddd (3.4, 3.4, 3.3, 3.3) |
| 4α | 2.32, dd (15.1, 3.9) | 2.36, dd (15.2, 3.9) | 2.16, dd (15.0, 2.6) | 2.18, dd (14.7, 3.1) |
| 4β | 1.62, ddd (14.5, 2.0, 2.0) b | 1.67, d (15.0) | 1.57, br d (14.6) | 1.47, ddd (14.8, 3.0, 3.0) b |
| 6α | 1.43, m | 1.45, m | 1.45, br d (14.4) | 1.49, m |
| 6β | 1.65, ddd (14.1, 13.8, 4.8) | 1.67, ddd (13.9, 13.9, 4.6) | 1.83 | 1.84, ddd (13.5, 13.5, 4.5) |
| 7α | 1.29, dddd (13.7, 13.7, 12.7, 3.3) | 1.32, m | 1.30, dddd (14.2, 14.2, 13.9, 4.4) | 1.30, m |
| 7β | 1.97, m | 2.01, m | 2.02, m | 2.00, m |
| 8 | 1.80, ddd (12.0, 11.7, 4.1) | 1.85, ddd (12.0, 12.0, 4.0) | 1.86 | 1.98, ddd (12.4, 12.4, 4.1) |
| 9 | 1.46, m | 1.54 | 1.76, dd (11.2, 11.2) | 1.77, dd (11.8, 10.3) |
| 11α | 1.51, m | 1.53 | ||
| 11β | 1.58, t (12.0) | 1.53 | 3.93, ddd (10.8, 10.8, 4.1) | 3.88, ddd (11.1, 11.0, 4.6) |
| 12α | 1.35, m | 1.38, dd (13.0, 13.0) | 1.53, dd (13.0, 11.5) | 1.53, dd (13.2, 11.6) |
| 12β | 1.50, m | 1.50, d (13.0) | 1.67, dd (13.4, 4.2) | 1.68, dd (13.4, 4.4) |
| 15α | 1.69 | 1.71 | 1.73 | 1.73 |
| 15β | 2.05, m | 2.07, m | 2.14, m | 2.13, m |
| 16α | 2.18, ddd (11.7, 9.4, 9.4) | 2.19, dddd (9.4, 9.1, 9.1, 3.6) | 2.20, m | 2.22, m |
| 16β | 1.72 | 1.73 | 1.76 | 1.75 |
| 17 | 2.54, dd (9.6, 6.1) | 2.55, dd (9.7, 6.0) | 2.62, dd (9.2, 6.5) | 2.61, dd (9.3, 6.3) |
| 18 | 0.74, s | 0.71, s | 0.76, s | 0.74, s |
| 19-a | 4.40, d (11.8) | 4.81, d (12.1) | 4.13, d (11.2) | 4.45, d (11.9) |
| 19-b | 4.06, d (11.6) | 4.62, d (11.8) | 3.84, d (11.3) | 4.42, d (11.9) |
| 21 | 7.42, dd (2.6, 1.1) | 7.42, dd (2.6, 1.0) | 7.45, dd (2.5, 0.9) | 7.45, dd (2.6, 1.1) |
| 22 | 7.98, dd (9.7, 2.6) | 7.98, dd (9.7, 2.6) | 7.94, dd (9.8, 2.5) | 7.96, dd (9.7, 2.6) |
| 23 | 6.28, dd (9.7, 1.1) | 6.28, dd (9.8, 1.0) | 6.28, dd (9.7, 0.8) | 6.28, dd (9.7, 1.0) |
| 19-COCH3 | 2.05, s | 2.07, s | ||
| Rha | ||||
| 1′ | 4.85, d (1.6) | |||
| 2′ | 3.78, dd (3.3, 1.7) | |||
| 3′ | 3.63, dd (9.5, 3.5) | |||
| 4′ | 3.40, dd (9.5, 9.5) | |||
| 5′ | 3.65, dq (9.5, 6.2) | |||
| 6′ | 1.26, d (6.3) |
| Position | 5 | 6 | 7 | 8 |
|---|---|---|---|---|
| 1α | 2.43, ddd (14.2, 3.4, 3.4) | 2.51, m | 5.24, br s | 6.52, dd (2.8, 2.8) |
| 1β | 2.07, ddd (14.1, 14.1, 3.8) | 1.75, m | ||
| 2α | 1.85, dddd (14.4, 14.4, 3.3, 3.3) | 1.78, m | 2.14, ddd (15.5, 3.4, 3.4) | 2.11, m |
| 2β | 1.64, ddd (14.4, 3.2, 3.2) | 1.62, ddd (13.7, 2.8, 2.8) | 2.04, br d (15.5) | 2.11, m |
| 3α | 4.08, dddd (2.8, 2.8, 2.8, 2.8) | 4.11, br s | 4.22, br s | 4.22, dddd (3.3, 3.3, 3.3, 3.3) |
| 4α | 2.10, dd (15.0, 3.1) | 2.11, dd (14.9, 2.8) | 2.27, dd (15.2, 3.7) | 2.25, dd (14.9, 3.5) |
| 4β | 1.45, ddd (14.8, 2.7, 2.7) b | 1.49, br d (13.7) | 1.68, ddd (14.9, 2.2, 2.2) b | 1.61, dd (14.9, 2.8) |
| 6α | 1.51, ddd (13.3, 3.6, 3.6) | 1.48, ddd (14.1, 3.7, 3.7) | 1.49, ddd (13.2, 3.9, 3.9) | 1.48, ddd (13.8, 3.8, 2.7) |
| 6β | 1.97, ddd (13.4, 13.4, 4.5) | 1.85, m | 1.77, ddd (13.6, 13.6, 4.6) | 1.80, ddd (13.7, 13.6, 4.6) |
| 7α | 1.34, dddd (13.6, 13.5, 13.5, 4.3) | 1.36, dddd (13.7, 13.7, 13.7, 4.7) | 1.36, dddd (14.0, 14.0, 13.9, 4.4) | 1.36, dddd (14.0, 14.0, 13.7, 4.3) |
| 7β | 2.10, dddd (13.6, 4.5, 4.4, 2.6) | 2.11, m | 2.13, dddd (13.5, 4.0, 4.0, 4.0) | 2.13, m |
| 8 | 2.42, ddd (12.4, 12.3, 4.2) | 2.43, ddd (12.4, 12.4, 4.2) | 2.38, ddd (12.3, 12.1, 4.0) | 2.49, ddd (12.4, 12.4, 4.0) |
| 9 | 1.80, dd (11.6, 11.6) | 1.83, dd (11.6, 11.6) | 1.67, dd (11.6, 11.6) | 1.68, dd (11.4, 11.4) |
| 11β | 4.71, d (11.2) | 4.61, d (11.2) | 4.65, d (11.1) | 5.07, d (10.9) |
| 15α | 1.32, m | 1.34, m | 1.29, m | 1.28, m |
| 15β | 1.73 | 1.75 | 1.75 | 1.72 |
| 16α | 2.00, dddd (10.0, 10.0, 9.6, 3.5) | 2.02, dddd (9.8, 9.8, 9.3, 4.7) | 2.01, dddd (9.6, 9.6, 9.5, 3.9) | 2.00, m |
| 16β | 1.73 | 1.74 | 1.74 | 1.75 |
| 17 | 4.11, dd (9.9, 7.0) | 4.12, dd (9.9, 7.0) | 4.11, dd (9.7, 6.9) | 4.11, dd (9.8, 6.8) |
| 18 | 0.92, s | 0.91, s | 0.91, s | 0.95, s |
| 19-a | 4.19, d (11.3) | 4.60, d (12.0) | 5.00, d (12.2) | 4.37, d (11.5) |
| 19-b | 4.01, d (11.3) | 4.50, d (12.0) | 4.85, d (12.0) | 4.29, d (11.6) |
| 21 | 7.52, dd (2.5, 0.9) | 7.53, dd (2.5, 0.8) | 7.52, dd (2.6, 0.6) | 7.52, br d (2.7) |
| 22 | 7.91, dd (9.7, 2.6) | 7.92, dd (9.7, 2.6) | 7.92, dd (9.8, 2.6) | 7.93, dd (9.7, 2.7) |
| 23 | 6.31, dd (9.7, 1.0) | 6.31, dd (9.7, 0.7) | 6.31, dd (9.7, 0.8) | 6.31, dd (9.7, 0.9) |
| 1-COCH3 | 2.04, s | |||
| 19-COCH3 | 2.08, s | 2.08, s |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moniuszko-Szajwaj, B.; Pecio, Ł.; Kowalczyk, M.; Stochmal, A. New Bufadienolides Isolated from the Roots of Kalanchoe daigremontiana (Crassulaceae). Molecules 2016, 21, 243. https://doi.org/10.3390/molecules21030243
Moniuszko-Szajwaj B, Pecio Ł, Kowalczyk M, Stochmal A. New Bufadienolides Isolated from the Roots of Kalanchoe daigremontiana (Crassulaceae). Molecules. 2016; 21(3):243. https://doi.org/10.3390/molecules21030243
Chicago/Turabian StyleMoniuszko-Szajwaj, Barbara, Łukasz Pecio, Mariusz Kowalczyk, and Anna Stochmal. 2016. "New Bufadienolides Isolated from the Roots of Kalanchoe daigremontiana (Crassulaceae)" Molecules 21, no. 3: 243. https://doi.org/10.3390/molecules21030243