
Alkaloids with Anti-Onchocercal Activity from Voacanga africana Stapf (Apocynaceae): Identification and Molecular Modeling


 ,
,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Isolation and Identification of Alkaloids
2.2. Anti-Onchocercal Activity
2.3. Molecular Modeling of Secondary Metabolites
2.3.1. Computation of Pharmacokinetic-Related Properties
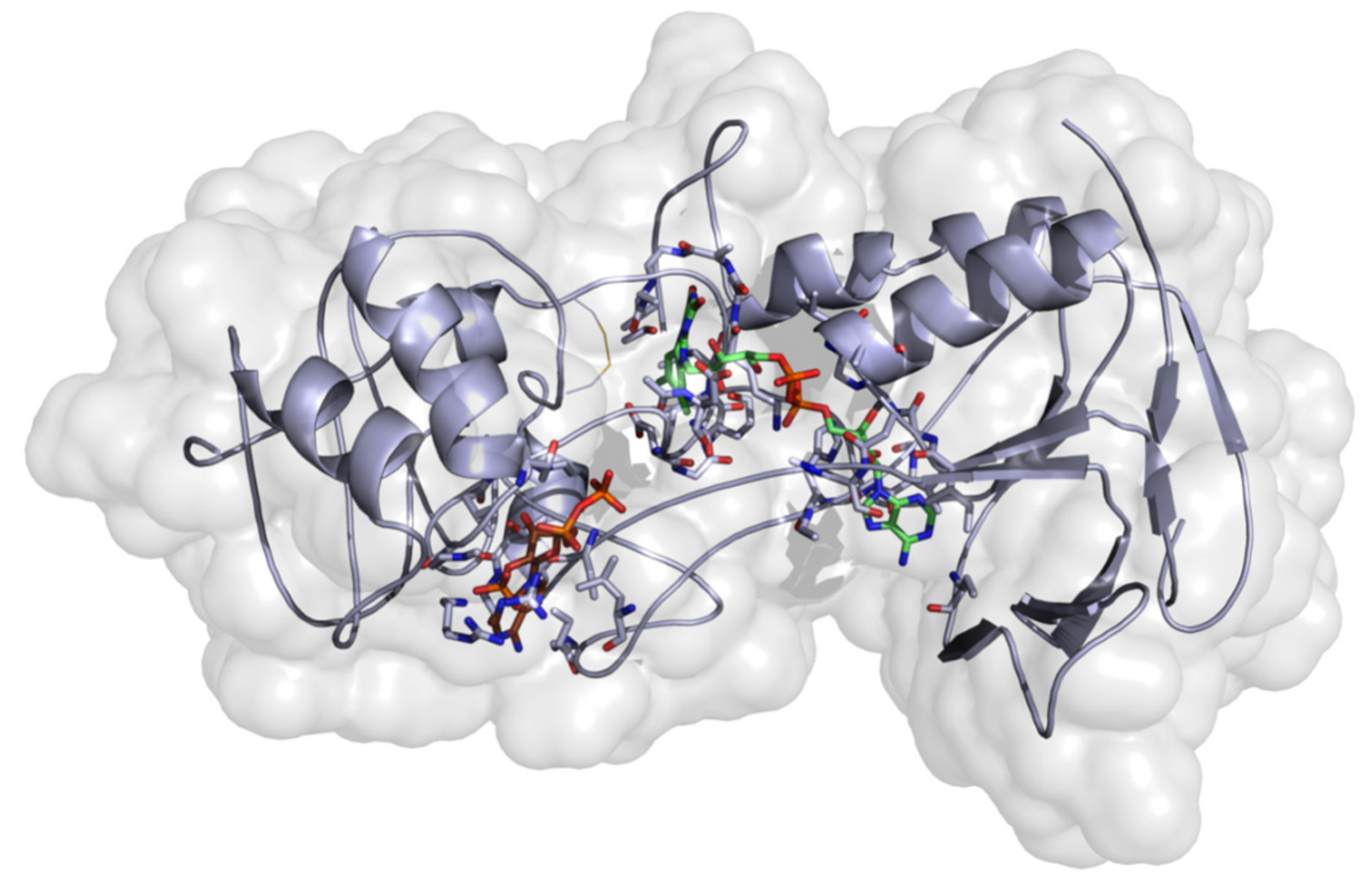
2.3.2. Homology Modeling
Model Development
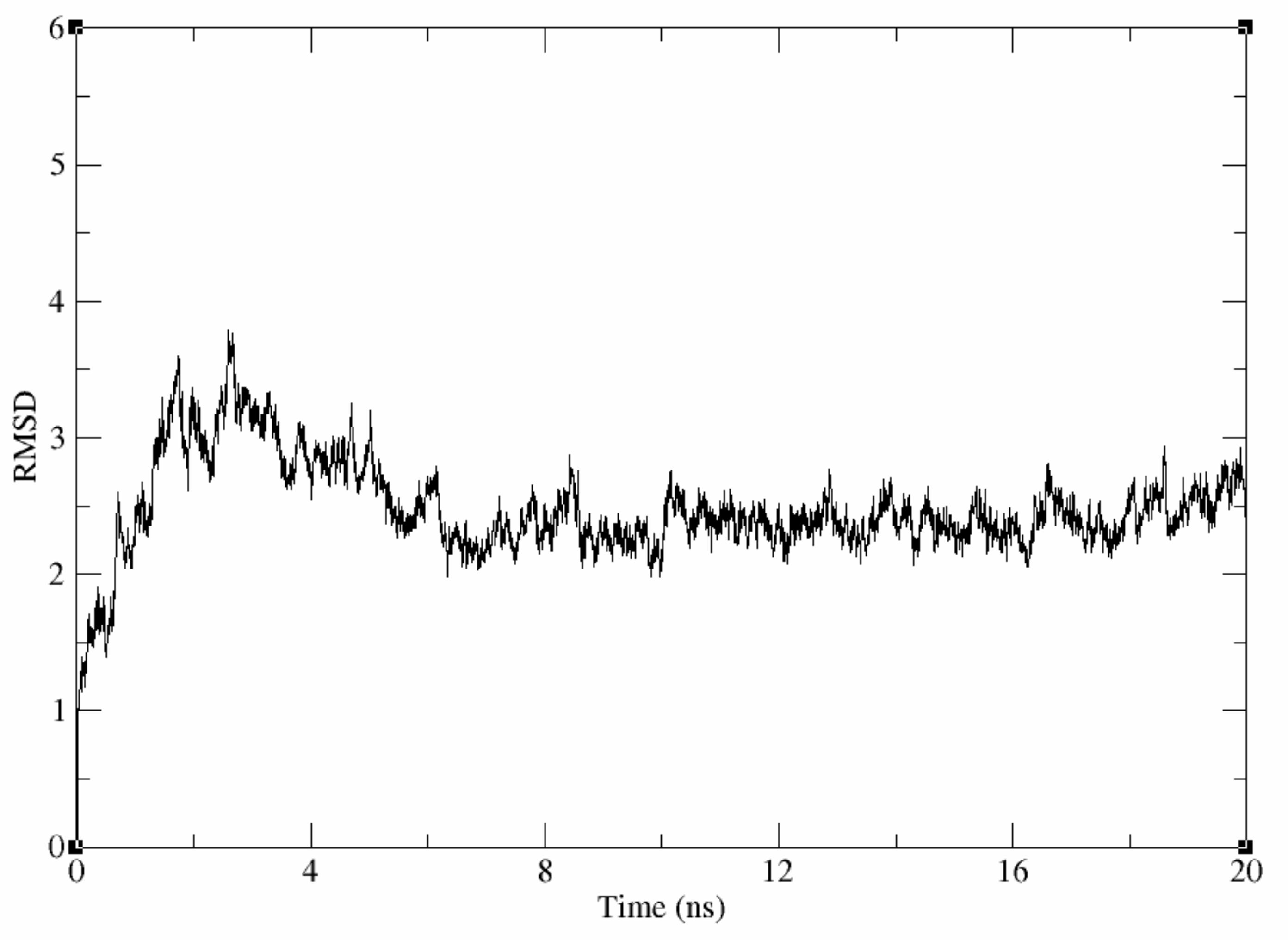
Evaluation of the Stability of the Generated Homology Model
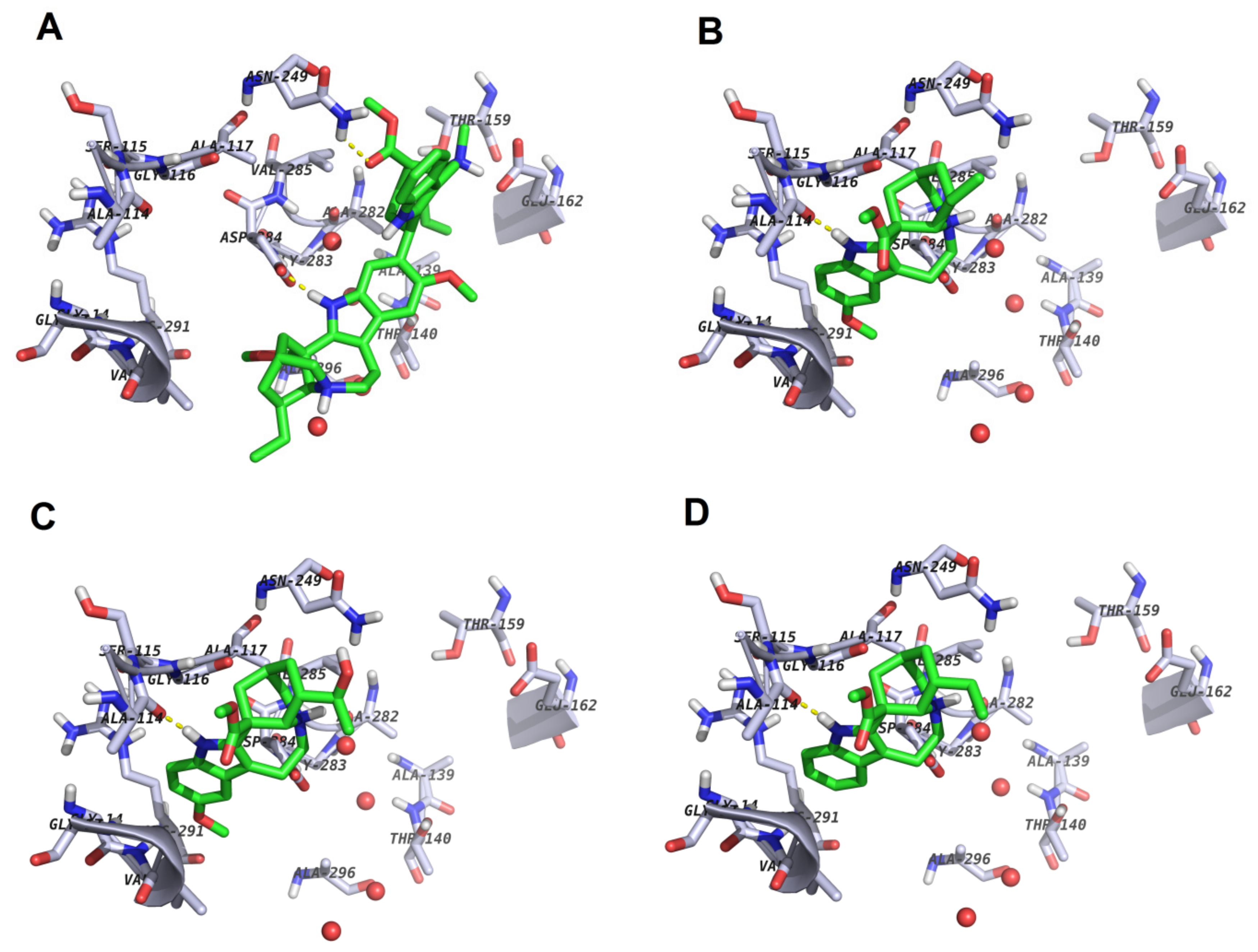
2.3.3. Docking
2.3.4. Binding Free Energy Calculations
2.3.5. Structure–Activity Relationships
- Compound 1 is more active than compound 2, being ~2 fold more active in both Mf and adult male worms. In our discussion, the activities against adult female worms were ignored, since the experimental activities were limited to a few dotted cases.
- The measured activities of the bisindoles (1, 7 and 8, possessing a vobasinyl unit) were much better than those of the mono indoles (2–4 and 6).
- Compound 5 was almost inactive in all assays and is not included in Table 3 and this discussion.
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. In Vitro Antimalarial Activity
3.4.1. Isolation of Onchocerca ochengi Adult Worms
3.4.2. Mammalian Cells for Microfilarial Cultures and Cytotoxicity Studies
3.4.3. Isolation and Culturing of Onchocerca ochengi Microfilariae
3.4.4. Preparation of Loa loa Microfilariae
3.4.5. Primary and Secondary Screens against Onchocerca ochengi Adult Worms
3.4.6. Primary and Secondary Screens against Onchocerca ochengi Microfilariae
3.4.7. Screens against Loa loa Microfilariae
3.4.8. Cytotoxicity Studies
3.4.9. Statistical Analysis
3.5. Molecular Modeling
3.5.1. Homology Modeling
Molecular Dynamic Simulation of the Generated Homology Model
3.5.2. Protein Preparation
3.5.3. Ligand Dataset Preparation
3.5.4. Ligand Docking and Scoring
3.5.5. Rescoring Docked Poses by Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tona, L.; Kambu, K.; Mesia, K.; Cimanga, K.; Apers, S.; De Bruyne, T.; Pieters, L.; Totte, J.; Vlietinck, A.J. Biological screening of traditional preparations from some medicinal plants used as antidiarrhoeal in Kinshasa, Congo. Phytomedicine 1999, 6, 59–66. [Google Scholar] [CrossRef]
- Liu, X.; Yanga, D.; Liu, J.; Ren, N. Synthesis and acetylcholinesterase inhibitory activities of tabersonine derivatives. Phytochem. Lett. 2015, 14, 17–22. [Google Scholar] [CrossRef]
- Chen, H.M.; Yang, Y.T.; Li, H.X.; Cao, Z.X.; Dan, X.M.; Mei, L.; Guo, D.L.; Song, C.X.; Dai, Y.; Hu, J.; et al. Cytotoxic monoterpenoid indole alkaloids isolated from the barks of Voacanga africana Staph. Nat. Prod. Res. 2016, 30, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.V.; Nyasse, B. Anti-ulcer compound from Voacanga africana with possible histamine H2 receptor blocking activity. Phytomedicine 2000, 7, 509–515. [Google Scholar] [CrossRef]
- Tan, P.V.; Penlap, V.B.; Nyasse, B.; Nguemo, J.D.B. Anti-ulceractions of the bark methanol extract of Voacanga africana in different experimental ulcer models in rats. J. Ethnopharmacol. 2000, 73, 423–428. [Google Scholar] [CrossRef]
- Jiofack, T.; Fokunang, C.; Kemeuze, V.; Fongnzossie, E.; Tsabang, N.; Nkuinkeu, R.; Mapongmetsem, P.M.; Nkongmeneck, B.A. Ethnobotany and phytopharmacopoea of the South-West ethnoecological region of Cameroon. J. Med. Plants Res. 2008, 2, 197–206. [Google Scholar]
- Hussain, H.; Hussain, J.; Al-Harrasi, A.; Green, I.R. Chemistry and biology of the genus Voacanga. Pharm. Biol. 2012, 50, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, M.; Iwai, M.; Kogure, N.; Kikura-Hanajiri, R.; Goda, Y.; Takayama, H. Aspidosperma-aspidosperma-type bisindole alkaloids from Voacanga africana. Tetrahedron 2013, 69, 796–801. [Google Scholar] [CrossRef]
- Dey, A.; Mukherjee, A.; Chaudhury, M. Alkaloids from Apocynaceae: Origin, pharmacotherapeutic properties, and structure-activity studies. Stud. Nat. Prod. Chem. 2017, 52, 373–487. [Google Scholar]
- Rao, K.V. Alkaloids of Voacanga africana, Stapf. I. voacafrine and voacafricine—two new alkaloids. J. Org. Chem. 1958, 23, 1455–1456. [Google Scholar] [CrossRef]
- Kitajima, M.; Iwai, M.; Kikura-Hanajiri, R.; Goda, Y.; Iida, M.; Yabushita, H.; Takayama, H. Discovery of indole alkaloids with cannabinoid CB1 receptor antagonistic activity. Bioorg. Med. Chem. 2011, 21, 1962–1964. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, M.; Takayama, H. Monoterpenoid bisindole alkaloids. Alkaloids 2016, 76, 259–310. [Google Scholar]
- Borakaeyabe, S.B.; Mbah, J.A.; Cho-Ngwa, F.; Metuge, J.A.; Efange, S.M.N. Isolation and characterization of filaricidal compounds from the stem bark of Voacanga africana, a plant used for onchocerciasis in Cameroon. J. Med. Plants Res. 2015, 9, 471–478. [Google Scholar]
- Trees, A.; Graham, S.; Renz, A.; Bianco, A.; Tanya, V. Onchocerca ochengi infections in cattle as a model for human onchocerciasis: Recent developments. Parasitology 2000, 120, 133–142. [Google Scholar] [CrossRef]
- Lobos, E.; Weiss, N.; Karam, M.; Taylor, H.R.; Ottesen, E.A.; Nutman, T.B. Animmunogenic Onchocerca volvulus antigen: A specific and early marker of infection. Science 1991, 251, 1603–1605. [Google Scholar] [CrossRef]
- Harada, M.; Asaba, K.N.; Iwai, M.; Kogure, N.; Kitajima, M.; Takayama, H. Asymmetric total synthesis of an iboga-type indole alkaloid, voacangalactone, newly isolated from Voacanga africana. Org. Lett. 2012, 14, 5800–5803. [Google Scholar] [CrossRef]
- Kutney, J.P.; Horinaka, A.; Ward, R.S.; Worth, B.R. Studies on the total synthesis of bisindole alkaloids within the voacamine family. Can. J. Chem. 1980, 58, 1829–1838. [Google Scholar] [CrossRef]
- Medeiros, W.L.B.; Vieira, I.J.C.; Mathias, L.; Braz-Filho, R.; Leal, K.Z.; Rodrigues-Filho, E.; Schripsema, J. Two known bis-indole alkaloids isolated from Tabernaemontana laeta: Complete 1H and 13C chemical shift assignments. Magn. Reson. Chem. 1999, 37, 676–681. [Google Scholar] [CrossRef]
- Nge, C.E.; Chong, K.W.; Thomas, N.F.; Lim, S.H.; Low, Y.Y.; Kam, T.S. Ibogan, a spidosperma, vincamine, and bisindole alkaloids from a Malayan Tabernaemontana corymbosa: Iboga alkaloids with C-20α substitution. J. Nat. Prod. 2016, 79, 1388–1399. [Google Scholar] [CrossRef]
- Sim, D.S.Y.; Teoh, W.Y.; Sim, K.S.; Lim, S.H.; Thomas, N.F.; Low, Y.Y.; Kam, T.S. Vobatensines A–F, cytotoxic iboga-vobasine bisindoles from Tabernaemontana corymbosa. J. Nat. Prod. 2016, 79, 1048–1055. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhu, W.-T.; Ding, X.; Huo, Z.-Q.; Donkor, P.O.; Adelakun, T.A.; Hao, X.-J.; Zhang, Y. Voacafrines A-N, aspidosperma-type monoterpenoid indole alkaloids from Voacanga africana with AChE inhibitory activity. Phytochemistry 2020, 181, 112566. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedula, V.S.P.; Sprague, S.; Schilling, J.K.; Kingston, D.G.I. New cytotoxic indole alkaloids from Tabernaemontana calcarea from the Madagascar rainforest. J. Nat. Prod. 2003, 66, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Cordell, G.A. Heyneanine hydroxyindolenine, a new indole alkaloid from Ervatamia coronaria var.plena. J. Nat. Prod. 1988, 51, 528–531. [Google Scholar] [CrossRef]
- Qu, Y.; Simonescu, R.; Luca, V.D. Monoterpene indole alkaloids from the fruit of Tabernaemontana litoralis and differential alkaloid composition in various fruit components. J. Nat. Prod. 2016, 79, 3143–3147. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.W.; Biemann, K. The hydroxyindolenine derivative of voacangine, a new indole alkaloid from Voacanga africana. Tetrahedron 1987, 24, 4223–4231. [Google Scholar] [CrossRef]
- Cho-Ngwa, F.; Abongwa, M.; Ngemenya, N.M.; Nyongbela, K.D. Selective activity of extracts of Margaritaria discoidea and Homalium africanum on Onchocerca ochengi. BMC Complement. Altern. Med. 2010, 10, 62. [Google Scholar] [CrossRef] [Green Version]
- Bulman, C.A.; Bidlow, C.M.; Lustigman, S.; Cho-Ngwa, F.; Williams, D.; Rascón, A.A., Jr.; Tricoche, N.; Samje, M.; Bell, A.; Suzuki, B.; et al. Repurposing auranofin as a lead candidate for treatment of lymphatic filariasis and onchocerciasis. PLoS Negl. Trop. Dis. 2015, 9, e0003534. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Ahmad, S. Role of solvent accessibility in structure based drug design. Curr. Comput. Aided Drug Des. 2005, 1, 223–235. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Cavalli, A.; Poluzzi, E.; De Ponti, F.; Recanatini, M. Toward a pharmacophore for drugs inducing the long QT syndrome: Insights from a CoMFA Study of HERG K+ channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar] [CrossRef]
- De Ponti, F.; Poluzzi, E.; Montanaro, N. Organising evidence on QT prolongation and occurrence of Torsades de Pointes with non antiarrhythmic drugs: A call for consensus. Eur. J. Clin. Pharmacol. 2001, 57, 185–209. [Google Scholar] [CrossRef] [PubMed]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Colmenarejo, G.; Alvarez-Pedraglio, A.; Lavandera, J.-L. Cheminformatic models to predict binding affinities to human serum albumin. J. Med. Chem. 2001, 44, 4370–4378. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016; Available online: https://ambermd.org/ (accessed on 12 November 2020).
- Cho-Ngwa, F.; Monya, E.; Azantsa, B.K.; Manfo, F.P.T.; Babiaka, S.B.; Mbah, J.A.; Samje, M. Filaricidal activities on Onchocerca ochengi and Loa loa, toxicity and phytochemical screening of extracts of Tragia benthami and Piper umbellatum. BMC Complement. Altern. Med. 2016, 16, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthley, E.G.; Schott, C.D. The toxicity of four concentrations of DMSO. Toxicol. Appl. Pharmacol. 1969, 15, 275–281. [Google Scholar] [CrossRef]
- The UniProt Consortium. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Costanzo, L.D.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE), 2016.08; Chemical Computing Group Inc., 1010 Sherbrooke St. West, Suite#910, Montreal, QC, Canada, H3A 2R7, 2016. Available online: https://www.chemcomp.com/Products.htm (accessed on 12 November 2020).
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Sagui, C.; Pedersen, L.G.; Darden, T.A. Towards an accurate representation of electrostatics in classical forcefields: Efficient implementation of multipolar interactions in biomolecular simulations. J. Chem. Phys. 2004, 120, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Toukmaji, A.; Sagui, C.; Board, J.; Darden, T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000, 113, 10913–10927. [Google Scholar] [CrossRef]
- Simoben, C.V.; Robaa, D.; Chakrabarti, A.; Schmidtkunz, K.; Marek, M.; Lancelot, J.; Kannan, S.; Melesina, J.; Shaik, T.B.; Pierce, R.J.; et al. A novel class of Schistosoma mansoni histone deacetylase 8 (HDAC8) inhibitors identified by structure-based virtual screening and in vitro testing. Molecules 2018, 23, 566. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated modeling program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [Green Version]
- Schrodinger. Schrödinger Release 2017–2: LigPrep; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Schrodinger. Schrödinger Release 2017-2: ConfGen; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple AMBER forcefields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | |||||||
|---|---|---|---|---|---|---|---|
| No. | δH | δC | δN | No. | δH | δC | δN |
| 1 | 7.74 (1H) | 127.2 | 1′ | 7.49 (1H) | 120.1 | ||
| 2 | 137.3 | 2′ | 137.2 | ||||
| 3 | 5.17 (1H) | 37.3 | 3′ | 2.89 (1H) 2.74 (1H) | 51.9 | ||
| 4 | 40.8 | 4′ | 20.3 | ||||
| 5 | 4.07 (1H) | 60.0 | 5′ | 3.40 (1H) 3.17 (1H) | 53.1 | ||
| 6 | 3.51 (1H) 3.28 (1H) | 19.5 | 6′ | 3.12 (1H) 2.99 (1H) | 22.2 | ||
| 7 | 110.0 | 7′ | 110.0 | ||||
| 8 | 129.9 | 8′ | 127.4 | ||||
| 9 | 7.57 (1H) | 117.4 | 9′ | 6.95 (1H, s) | 99.2 | ||
| 10 | 7.08 (1H) | 119.0 | 10′ | 150.9 | |||
| 11 | 7.07 (1H) | 121.6 | 11′ | 129.8 | |||
| 12 | 7.07 (1H) | 109.8 | 12′ | 6.76 (1H) | 110.3 | ||
| 13 | 135.8 | 13′ | 130.3 | ||||
| 14 | 2.58 (1H) 2.02 (1H) | 36.3 | 14′ | 1.82 (1H) | 27.3 | ||
| 15 | 3.80 (1H) | 33.6 | 15′ | 1.70 (1H) 1.11 (1H) | 32.0 | ||
| 16 | 2.75 (1H) | 47.0 | 16′ | 54.9 | |||
| 17 | 17′ | 2.50 (1H) 1.78 (1H) | 36.4 | ||||
| 18 | 1.69 (3H) | 12.3 | 18′ | 0.90 (3H) | 11.6 | ||
| 19 | 5.36 (1H) | 118.8 | 19′ | 1.56 (1H) 1.45 (1H) | 26.7 | ||
| 20 | 137.8 | 20′ | 1.31 (1H) | 39.0 | |||
| 21 | 3.76 (1H) 2.95 (1H) | 52.6 | 21′ | 3.53 (1H) | 57.2 | ||
| 22 CO | 171.5 | 22′ CO | 175.3 | ||||
| 22 OMe | 2.48 (3H) | 49.9 | 22′ OMe | 3.67 (3H) | 52.4 | ||
| 4 NMe | 2.63 (3H) | 42.4 | 10′OMe | 4.03 (3H) | 56.1 | ||
| * Compounds Tested (at 30 μM) | % Mf Motility Reduction after 24 h | % Adult Male Worm Motility Reduction after 24 h | % Adult Female Worm Death after 120 h |
|---|---|---|---|
| 1 | 100 | 100 | 65 |
| 2 | 100 | 100 | 100 |
| 3 | 100 | 100 | 50 |
| 4 | 100 | 100 | 50 |
| 5 | 0 | 0 | 0 |
| 6 | 100 | 100 | 50 |
| 7 | 100 | 100 | 65 |
| 8 | 100 | 100 | 50 |
| 9 | 0 | 0 | 0 |
| Ivermectin (10 μg/mL) | 100 | NA | NA |
| Auranofin (10 μM) | 100 | 100 | 100 |
| 2% DMSO | 0 | 0 | 0 |
| Mf | Adult Male Worm | Adult Female Worm | Monkey Kidney Cells (LLC-MK2) | Mf | Adult Male Worm | Adult Female Worm | Monkey Kidney Cells (LLC-MK2) | |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | |||||||
| IC50 (μM) | 3.69 | 4.45 | - | ≥30 | 5.49 | 9.07 | - | ≥30 |
| IC100 (μM) | 7.38 | 8.90 | >30 | - | 10 | 20 | >30 | - |
| SI | 8.13 | 6.74 | 5.46 | 3.30 | 2.83 | |||
| 3 | 4 | |||||||
| IC50 (μM) | 4.34 | 8.07 | - | ≥30 | 4.21 | 8.68 | - | ≥30 |
| IC100 (μM) | 8.68 | 16.14 | >30 | - | 8.42 | 17.36 | >30 | - |
| SI | 6.91 | 3.71 | 7.13 | 3.45 | ||||
| 6 | 7 | |||||||
| IC50 (μM) | 4.72 | 9.07 | - | ≥30 | 2.49 | 3.45 | - | ≥30 |
| IC100 (μM) | 9.44 | 18.14 | >30 | - | 10 | 10 | >30 | - |
| SI | 6.35 | 3.30 | ||||||
| 8 | ||||||||
| IC50 (μM) | 2.49 | 3.45 | - | ≥30 | ||||
| IC100 (μM) | 4.98 | 6.90 | >30 | - | ||||
| SI | 12.04 | 8.69 | ||||||
| Metabolite | a #stars | b CNS | c MW (Da) | d SASA | e FOSA | f FISA | g Volume |
|---|---|---|---|---|---|---|---|
| 1 | 9 * | 1 | 706.9 | 1067.5 * | 786.3 * | 80.0 | 2140.6 * |
| 2 | 0 | 2 | 368.5 | 626.1 | 480.8 | 28.0 | 1171.6 |
| 3 | 0 | 1 | 384.5 | 642.5 | 461.5 | 64.5 | 1191.3 |
| 4 | 0 | 2 | 338.4 | 588.5 | 387.1 | 28.0 | 1094.4 |
| 5 | 0 | 2 | 368.5 | 635.5 | 485.4 | 31.7 | 1175.4 |
| 6 | 0 | 1 | 384.5 | 642.4 | 461.5 | 64.5 | 1191.3 |
| 7 | 9 * | 1 | 706.9 | 1057.7 * | 781.6 * | 71.7 | 2130.5 * |
| 8 | 10 * | 1 | 722.9 | 1091.1 * | 769.0 * | 119.3 | 2156.9 * |
| 9 | 10 * | 1 | 722.9 | 1089.4 * | 768.4 * | 115.5 | 2163.6 * |
| Metabolite | h HBD | i HBA | j logP | k logS | l logHERG | m Caco-2 | n logBB |
| 1 | 1 | 9 | 7.2 * | −8.0 * | −8.66 * | 26.8 | 0.2 |
| 2 | 0 | 4 | 4.5 | −4.4 | −5.11 | 1340.2 | 0.5 |
| 3 | 1 | 5 | 3.7 | −4.1 | −5.31 | 604.0 | 0.1 |
| 4 | 0 | 3 | 4.5 | −4.3 | −5.15 | 1341.8 | 0.6 |
| 5 | 0 | 4 | 4.5 | −4.6 | −5.29 | 1237.7 | 0.5 |
| 6 | 1 | 5 | 3.7 | −4.1 | −5.31 | 604.0 | 0.1 |
| 7 | 1 | 9 | 7.2 * | −7.8 * | −8.62 * | 32.1 | 0.3 |
| 8 | 2 | 10 | 6.2 | −7.5 * | −8.89 * | 11.4 | −0.4 |
| 9 | 2 | 10 | 6.2 | −7.5 * | −8.86 * | 12.4 | −0.4 |
| Metabolite | o MDCK | p logKp | q #metab | r logKHSA | s PHOA | t Ro5 | u Ro3 |
| 1 | 70.4 | −6.1 | 10 * | 2.49 * | 92.7 | 2 * | 2 * |
| 2 | 282.3 | −3.4 | 2 | 0.89 | 50.3 | 0 | 0 |
| 3 | 133.9 | −4.0 | 3 | 0.63 | 69.0 | 0 | 0 |
| 4 | 268.8 | −3.3 | 1 | 0.91 | 42.0 | 0 | 0 |
| 5 | 836.0 | −3.5 | 2 | 0.90 | 50.1 | 0 | 0 |
| 6 | 340.4 | −4.0 | 3 | 0.63 | 69.1 | 0 | 0 |
| 7 | 24.1 * | −5.9 | 10 * | 2.47 * | 95.1 | 2 * | 2 * |
| 8 | 9.2 * | −6.7 | 10 * | 2.09 * | 118.8 | 2 * | 3 * |
| 9 | 26.4 | −6.6 | 10 * | 2.11 * | 118.1 | 2 * | 3 * |
| Compound | Minimization Energy (Amber12, kcal/mol) | Docking Score (SP *, kcal/mol) |
|---|---|---|
| 1 | −30.48 | −4.25 |
| 1aa | −32.02 | −5.07 |
| 1bb | −32.33 | −4.89 |
| 2 | −32.01 | −5.07 |
| 3 | −28.94 | −5.26 |
| 4 | −26.79 | −4.54 |
| 5 | −28.72 | −4.80 |
| 6 | −28.88 | −5.26 |
| 7 | −30.43 | −4.25 |
| 7aa | −31.99 | −5.08 |
| 7bb | −21.83 | −4.18 |
| 8 | −28.41 | −4.73 |
| 8aa | −30.34 | −5.28 |
| 8bb | −21.91 | −4.18 |
| 9 | −40.53 | −5.20 |
| 9aa | −29.65 | −5.03 |
| 9bb | −21.59 | −4.18 |
| Auranofin | −47.38 | −6.37 |
| Compound | ΔGbind | Eele | Evdw | Esol |
|---|---|---|---|---|
| 1 | −38.59 | −94.21 | −33.61 | 89.23 |
| 1a | −23.65 | −92.54 | −18.71 | 87.6 |
| 2 | −33.95 | −75.89 | −29.02 | 70.96 |
| 3 | −34.01 | −68.58 | −29.51 | 64.08 |
| 4 | −29.32 | −70.64 | −25.32 | 66.64 |
| 5 | −26.57 | −111.25 | −23.41 | 108.08 |
| 6 | −27.74 | −63.78 | −22.78 | 58.82 |
| 7 | −40.47 | −81.75 | −36.67 | 77.95 |
| 7a | −24.67 | −39.34 | −21.48 | 36.15 |
| 9 | −29.83 | −155.19 | −24.87 | 150.22 |
| 9b | −28.49 | −56.25 | −23.83 | 51.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babiaka, S.B.; Simoben, C.V.; Abuga, K.O.; Mbah, J.A.; Karpoormath, R.; Ongarora, D.; Mugo, H.; Monya, E.; Cho-Ngwa, F.; Sippl, W.; et al. Alkaloids with Anti-Onchocercal Activity from Voacanga africana Stapf (Apocynaceae): Identification and Molecular Modeling. Molecules 2021, 26, 70. https://doi.org/10.3390/molecules26010070
Babiaka SB, Simoben CV, Abuga KO, Mbah JA, Karpoormath R, Ongarora D, Mugo H, Monya E, Cho-Ngwa F, Sippl W, et al. Alkaloids with Anti-Onchocercal Activity from Voacanga africana Stapf (Apocynaceae): Identification and Molecular Modeling. Molecules. 2021; 26(1):70. https://doi.org/10.3390/molecules26010070
Chicago/Turabian StyleBabiaka, Smith B., Conrad V. Simoben, Kennedy O. Abuga, James A. Mbah, Rajshekhar Karpoormath, Dennis Ongarora, Hannington Mugo, Elvis Monya, Fidelis Cho-Ngwa, Wolfgang Sippl, and et al. 2021. "Alkaloids with Anti-Onchocercal Activity from Voacanga africana Stapf (Apocynaceae): Identification and Molecular Modeling" Molecules 26, no. 1: 70. https://doi.org/10.3390/molecules26010070